Причины возникновения
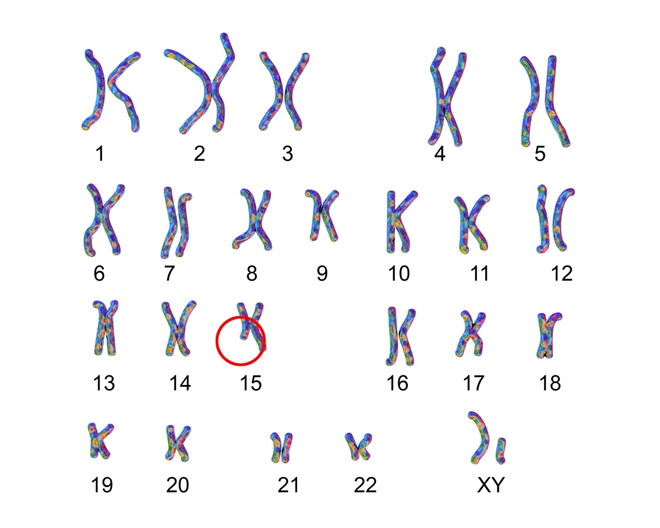
Выявить однозначные факторы возникновения патологии у детей невозможно. В большинстве случаев определяется изменение состава 15 хромосомы, но дефекты в ней носят различный характер. У 10% пациентов хромосомные аномалии не выявляются.

Возможные изменения в 15 хромосоме:
- делеция, т.е. потеря части генетического материала. При синдроме Ангельмана часто определяют делецию 15q12. Этот участок связан с активацией большого числа генов, принимающих участие в развитии нервной системы и внутренних органов;
- в 3–5% случаев патологии генетики обнаруживают однородительскую дисомию. Это состояние, при котором в кариотипе ребенка имеются две отцовских 15 хромосомы. Материнская копия при этом отсутствует;
- во внутриутробном периоде развития важную роль играет локус UBE3A, локализованный на 15 хромосоме. При его эпигенетическом «выключении» развиваются симптомы заболевания;
- мутация в локусе UBE3A выявляется у 5–10% больных детей.
Больные с патологией и их родители нуждаются в консультации врача-генетика. Специалист проводит молекулярно-генетические исследования, направленные на выявление причины появления синдрома Ангельмана.
Клинические проявления
Синдром Ангельмана - патология, обусловленная генетической аномалией, имеет и признаки нарушения психического развития, и внешние признаки. У детей с синдромом Ангельмана характерный внешний вид – непроизвольная, неестественная улыбка, частый смех, хаотические движения рук. Потому болезнь имеет и другие названия – «синдром Петрушки» или «синдром счастливой куклы».
У детей с патологией встречаются различные клинические проявления. Первые симптомы выявляют в возрасте от 6 до 12 месяцев, когда становится видно отставание ребенка в психомоторном развитии. В зависимости от распространенности признаки делят на 3 группы:
- встречаются у всех больных: тяжелые нарушения развития интеллектуальной и двигательной сферы и отклонения в поведении. Дети без причины смеются, улыбаются, отличаются повышенной возбудимостью и не могут сконцентрироваться на одном занятии. Кроме того, к данной группе симптомов относят двигательные нарушения, тремор, а также плохо развитую речь;
- клинические проявления, встречающиеся у 80% пациентов: микроцефалия, судорожные приступы, аномальная активность головного мозга на ЭЭГ в виде волн с высокой амплитудой;
- симптомы, отмечающиеся у меньшинства больных: сниженная пигментация радужки и кожного покрова, различные варианты косоглазия, нарушения глотания и сосания, повышение сухожильных рефлексов, бессонница, постоянная жажда и др.
Болезнь Ангельмана имеет разнообразную клиническую картину, что делает ее похожей на синдром Эдвардса и другие хромосомные аномалии. В связи с этим, родители не должны самостоятельно диагностировать заболевание. Им следует обращаться за профессиональной медицинской помощью.
Диагностические мероприятия

Диагностика заболевания может быть проведена в период беременности. С этой целью применяют инвазивные и неинвазивные методики. Инвазивные процедуры применяются в медицине дольше, однако их проведение сопряжено с определенным риском для здоровья плода и беременной. Они заключаются в исследовании крови ребенка или материала плаценты, полученных через матку.
Современный метод диагностики – исследование плодной ДНК, циркулирующей в крови матери. При этом специалисты способны оценить генетический материал и выявить его аномалии, характерные не только для Ангельмана, но и для патологии Патау и пр. К хромосомным заболеваниям относят синдром Прадера–Вилли, характеризующийся потерей части 15 хромосомы. Комплексное генетическое обследование позволяет проводить дифференциальную диагностику между врожденными патологиями.
У большинства больных синдром выявляют в возрасте от 2 до 5 лет. Родители обращают внимание на задержку развития ребенка, плохо сформированную речь и внешний вид. При обращении к специалисту, назначаются генетические исследования с определением кариотипа и отдельных мутаций. Дополнительно оценивают состояние внутренних органов и головного мозга с помощью УЗИ, компьютерной томографии и других методов.
Подходы к лечению
Синдром Ангельмана считается неизлечимой патологией, так как восстановление нормального строения хромосом невозможно. Лечение основывается на симптоматическом подходе и направлено на устранение отдельных клинических проявлений. Это позволяет повысить качество жизни ребенка и уменьшить риск развития негативных последствий болезни. К методам терапии относят:
- противоэпилептическое лечение с применением антиконвульсантов. Терапия показана больным, имеющим судорожные припадки. Предпочтение отдается медикаментам на основе вальпроевой кислоты;
- регулярные занятия лечебной физкультурой. ЛФК позволяет развить мелкую моторику, улучшить походку и двигательную активность детей. Занятия проводятся специалистами по лечебной физкультуре и родителями;
- пациентов с болезнью обучают языку жестов. Это необходимо для их нормальной социализации в обществе. Врачи рекомендуют проводить обучение в раннем возрасте, не дожидаясь взросления ребенка;
- когнитивно-поведенческая терапия снижает уровень возбудимости, тревожности и улучшает способность детей концентрировать внимание. Благодаря регулярным занятиям с психотерапевтом ребенок уверенно чувствует себя в коллективе и способен общаться со сверстниками.
При подборе лечения врачи учитывают индивидуальные особенности больного и выраженность у него клинических проявлений заболевания. Самолечение при патологии противопоказано, так как эффективная терапия возможна только при комплексном и профессиональном подходе.
Также интересно почитать: синдром вилли прадера
Фото: Depositphotos

